

-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
Genome Biology |单细胞组学与疾病研究中心郑小琪团队开发空间转录组数...
【字体: 大 中 小 】 时间:2024年11月13日 来源:上海交通大学医学院
编辑推荐:
近日,上海交通大学公共卫生学院、上海交通大学医学院单细胞组学与疾病研究中心郑小琪课题组在基因组学领域顶级期刊Genome Biology杂志发表了题为GraphPCA: a fast and interpretable dimension reduction algorithm for spatial transcriptomics data的方法论文章

近日,上海交通大学公共卫生学院、上海交通大学医学院单细胞组学与疾病研究中心郑小琪课题组在基因组学领域顶级期刊Genome Biology杂志发表了题为GraphPCA: a fast and interpretable dimension reduction algorithm for spatial transcriptomics data的方法论文章。该研究开发了一个快速、可解释性的拟线性降维算法——GraphPCA。基于模拟实验及真实数据的评估结果表明,GraphPCA有效提升了包含空间域检测、降噪以及轨迹推断等多项下游分析任务的性能。这项研究为空间转录组数据的分析提供了一个强有力的新工具,有助于更深入地理解细胞在组织中的复杂相互作用和功能。

近年来,空间转录组(Spatial transcriptomics, ST) 技术的快速发展使得研究人员在获取基因表达谱的同时保留了细胞在组织中的空间位置信息,从而能够揭示组织内细胞在空间结构上的异质性。然而,空间转录组数据具有高稀疏性、高维性和低信噪比等特点,为后续数据分析带来了巨大挑战。作为下游分析前必要的预处理步骤,降维不仅可以提高信噪比,还能有效缓解维数灾难。目前,大多数研究人员直接将针对单细胞转录组数据的降维算法应用到空间转录组数据(如Seurat、Scanpy、STUtility等)。然而,这些方法未能充分利用ST数据中的空间信息,可能导致低维嵌入的效率降低,甚至错误的生物学发现。虽然近期已有一些专门针对ST数据的降维算法(如SpatialPCA、DR-SC等),但这些方法或依赖复杂的参数推断,或缺乏模型可解释性。为了克服这些缺陷,本研究基于图正则化表示以及主成分分析法,开发了一种快速、可解释的拟线性降维算法——GraphPCA,该算法可以有效处理空间转录组数据,并提高了低维嵌入的生物学解释能力。
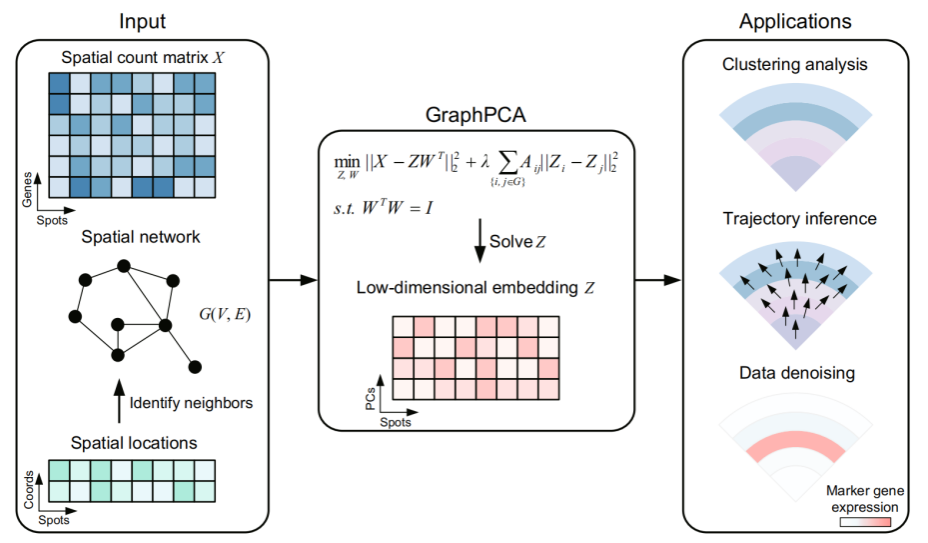
GraphPCA建立在灵活的主成分分析框架上,它通过利用位点/细胞之间的空间邻域结构作为图约束,使得低维嵌入能够有效地保留位置信息。GraphPCA的输入包括基因表达矩阵和位点的空间坐标,这些信息被用于构建位点/细胞间的空间邻域图(默认为k近邻图)。与经典的主成分分析方法不同,GraphPCA通过求解一个受空间邻域图约束的优化问题来推断整合了空间位置和基因表达信息的低维嵌入矩阵。由于该优化问题存在封闭解,GraphPCA的计算效率远高于基于深度学习的方法,从而可以高效地处理不同规模的ST数据。通过图约束,GraphPCA可以使相邻位点/细胞在低维空间中的投影更加接近,并且每个嵌入维度都与特定的空间基因表达模式高度相关,这使得基因-成分的投影矩阵能够反映共表达基因模块的空间表达差异性。
作者随后在大量模拟数据和不同物种、组织区域、测序技术的真实数据上进行了广泛的评估,验证了GraphPCA得到的低维嵌入在空间域检测、轨迹推断和去噪等下游分析任务中的性能。此外,GraphPCA模型的灵活性使其能够轻松扩展到多样本整合,通过融合其它切片的基因表达信息进一步提高空间域检测的准确性。
上海交通大学公共卫生学院、上海交通大学医学院单细胞组学与疾病研究中心的郑小琪教授为该论文的通讯作者,中心科研助理杨纪元为该论文的第一作者,上海交通大学自然科学研究院的刘林副教授为课题的开展提供了大力支持。该研究得到国家自然科学基金、上海市科技创新行动计划自然科学基金及数据科学与智慧教育教育部重点实验室的经费支持。
下载安捷伦电子书《通过细胞代谢揭示新的药物靶点》探索如何通过代谢分析促进您的药物发现研究
10x Genomics新品Visium HD 开启单细胞分辨率的全转录组空间分析!
生物通微信公众号


知名企业招聘
今日动态 | 人才市场 | 新技术专栏 | 中国科学人 | 云展台 | BioHot | 云讲堂直播 | 会展中心 | 特价专栏 | 技术快讯 | 免费试用
版权所有 生物通
Copyright© eBiotrade.com, All Rights Reserved
联系信箱:
粤ICP备09063491号












